Hemophilia: History, Symptoms, Treatment Options, and Drug Advances

Hemophilia: Origin, History, Symptoms, Treatments, and Drug Development
Hemophilia is a rare, inherited bleeding disorder in which the blood doesn’t clot properly, leading to excessive bleeding after an injury or surgery and sometimes spontaneous bleeding into joints or organs. It primarily affects males due to its X-linked genetic inheritance pattern, though females can also be carriers.
Origin and Historical Background
The origins of hemophilia are ancient. Historical records suggest that similar conditions were recognized as early as the 2nd century in ancient texts, notably in Jewish Talmudic law. There, boys who had two brothers die from bleeding after circumcision were excused from the ritual due to their risk of excessive bleeding, illustrating an early awareness of a hereditary bleeding disorder.
The term "hemophilia" itself came into use in the 19th century. It was coined in 1828 by Friedrich Hopff, a Swiss physician, based on the Greek words "haima" (blood) and "philia" (love), which together mean "a love of blood," or a propensity for bleeding. The condition became known as the "Royal Disease" in the 19th century due to its prominence in the royal families of Europe, notably in the descendants of Queen Victoria, who was a carrier. Her descendants, including her son Leopold and several grandsons, suffered from hemophilia, which subsequently spread throughout European royal families due to intermarriage.
Types of Hemophilia
Hemophilia is categorized into two primary types based on the deficient clotting factor:
Hemophilia A:
Caused by a deficiency in factor VIII, this is the most common type, representing approximately 80% of cases.
Hemophilia B:
Also known as "Christmas disease" (named after Stephen Christmas, the first known patient), it is caused by a deficiency in factor IX.
There is also a rarer type known as Hemophilia C, resulting from a deficiency in factor XI, though this is usually less severe and occurs in both males and females.
Symptoms of Hemophilia
Symptoms of hemophilia can vary from mild to severe, depending on the level of clotting factor in the blood. Common symptoms include:
1. Uncontrolled Bleeding: This can occur following an injury, surgery, or spontaneously without an apparent cause.
2. Prolonged Bleeding: Patients may experience prolonged bleeding even from minor cuts.
3. Internal Bleeding: This is particularly problematic in joints like knees, elbows, and ankles, causing pain, swelling, and stiffness.
4. Bruising: Frequent and extensive bruising can occur, especially in severe cases.
5. Blood in Urine or Stool: This is due to bleeding in the gastrointestinal or urinary tract.
6. Nosebleeds and Gum Bleeds: These are common but can be difficult to control.
In severe cases, hemophiliacs may experience life-threatening bleeds in vital organs, particularly the brain, which can lead to seizures, paralysis, or even death if untreated.
Treatment Processes
There is currently no cure for hemophilia, but treatments have improved significantly over the past century, allowing patients to lead relatively normal lives.
1. Replacement Therapy:
The primary treatment involves replacing the missing clotting factors. This can be done with:
Plasma-derived Concentrates:
Initially, these were derived from human blood plasma, but due to contamination risks (notably with HIV and hepatitis C in the 1970s and 1980s), they are now carefully screened.
Recombinant Factor Concentrates:
These synthetic factors (manufactured using genetic engineering techniques) are now the gold standard for hemophilia treatment as they pose no risk of viral transmission.
On-demand Therapy:
For minor bleeds, factor replacement is provided as needed.
Prophylactic Therapy:
Routine factor replacement can prevent bleeds, especially in children and those with severe hemophilia.
2. Desmopressin (DDAVP):
For some cases of mild Hemophilia A, desmopressin, a synthetic hormone, can help increase the release of factor VIII from storage in the body. This is not effective for Hemophilia B.
3. Antifibrinolytic Therapy:
Drugs like tranexamic acid and aminocaproic acid prevent the breakdown of clots, supporting clot stability and reducing bleeding risk. These are often used alongside replacement therapy or for minor bleeds.
4. Gene Therapy:
This innovative approach, still largely experimental, aims to correct the underlying genetic defect. Trials have shown promising results, with some patients achieving sustained levels of clotting factors. It remains an area of active research.
Development of Hemophilia Drugs
Drug development for hemophilia has progressed from basic blood transfusions to advanced genetic engineering:
Blood and Plasma Transfusions:
Before the 1960s, patients relied on whole blood or plasma transfusions, which were only marginally effective. This was due to the low concentration of clotting factors in plasma and challenges with clotting time control.
Factor Concentrates (1960s):
In the 1960s, scientists developed the ability to concentrate clotting factors from plasma, making treatment more effective and accessible.
However, in the 1980s, contaminated blood supplies led to HIV and hepatitis C infections among hemophiliacs, a tragic period that spurred development for synthetic options.
Recombinant Factor Replacement (1990s):
In the early 1990s, recombinant DNA technology allowed for the production of synthetic clotting factors. This eliminated the risk of viral contamination and improved the safety of treatment. Today, these are the primary choice for most patients.
Extended Half-Life Factor Products (2010s):
Recent developments have focused on prolonging the half-life of factor products, reducing the frequency of infusions. PEGylation and fusion technologies have allowed for factors that can last longer in the bloodstream, improving patient compliance and quality of life.
Gene Therapy (Present and Future):
Gene therapy, still experimental, has shown promising results in early trials. By delivering functional copies of defective genes directly into patients, this approach may offer long-term or even permanent restoration of clotting factors.
Hemophilia has progressed from a mysterious and often fatal condition to one that can be effectively managed with modern medicine. Today, a combination of replacement therapy, prophylactic infusions, and the possibility of gene therapy are transforming the outlook for hemophiliacs, enabling them to live longer and more active lives. Although gene therapy offers hope for a permanent cure, it remains out of reach for most patients.
Key Drugs Used in Hemophilia Treatment
Several drugs are used to manage hemophilia, primarily by replacing or mimicking the missing clotting factors (factor VIII for hemophilia A and factor IX for hemophilia B).
Some drugs also help to prevent the breakdown of clots, while others stimulate the release of clotting factors from the body. Here are some of the key drugs used in hemophilia treatment:
1. Factor VIII Replacement Therapy (for Hemophilia A)
Factor VIII replacement therapy is the primary treatment for hemophilia A. These medications contain factor VIII, the clotting protein that patients with hemophilia A lack.
Advate:
A recombinant (synthetic) factor VIII product, Advate is commonly used to prevent or control bleeding episodes. It’s derived from genetically engineered cells and is administered via intravenous injection.
Kovaltry:
Another recombinant factor VIII, Kovaltry offers flexibility in dosing and can be used for both on-demand and prophylactic treatment. It is especially noted for its stability and consistency in managing hemophilia A.
Eloctate:
Eloctate is an extended half-life (EHL) factor VIII product, which means it stays in the bloodstream longer, allowing for less frequent dosing. Eloctate is used primarily for preventive therapy.
Hemlibra (Emicizumab):
Unlike traditional factor replacement therapies, Hemlibra is a monoclonal antibody that mimics the function of factor VIII. It is administered subcutaneously (under the skin) and is often used for patients with hemophilia A who have developed inhibitors against factor VIII.
2. Factor IX Replacement Therapy (for Hemophilia B)
For hemophilia B, factor IX replacement products are used to manage bleeding.
BeneFIX:
This recombinant factor IX product is used for both on-demand treatment of bleeding episodes and prophylactic therapy in patients with hemophilia B.
Alprolix:
Alprolix is an extended half-life factor IX therapy that allows for less frequent infusions due to its longer duration in the bloodstream.
Idelvion:
Another extended half-life product, Idelvion is designed to provide effective control with fewer doses, making it convenient for prophylactic use. Its longevity in the bloodstream is due to fusion with albumin, a naturally occurring protein that helps maintain its presence.
Rebinyn:
Rebinyn is a recombinant factor IX with a modification that extends its half-life, allowing it to stay longer in the bloodstream and requiring less frequent dosing.
3. Desmopressin (DDAVP)
DDAVP (Stimate):
Desmopressin is a synthetic hormone that helps release stored factor VIII from blood vessels. It’s typically used for mild hemophilia A cases and is effective in raising factor VIII levels in some patients. It is administered as a nasal spray (Stimate) or through an injection. However, it is not effective for hemophilia B.
4. Antifibrinolytic Agents
Antifibrinolytic drugs help to prevent the breakdown of blood clots, supporting clot stability and reducing bleeding. They are often used in combination with factor replacement therapies, especially for mucosal bleeding, such as nosebleeds and dental procedures.
Tranexamic Acid (Cyklokapron, Lysteda):
Tranexamic acid prevents the breakdown of clots by inhibiting enzymes that dissolve blood clots (fibrinolysis). It is available in both oral and injectable forms and is often used alongside other therapies.
Aminocaproic Acid (Amicar):
Aminocaproic acid functions similarly to tranexamic acid by preventing fibrinolysis and supporting clot stability. It is usually administered orally and is effective for mucosal bleeds.
5. Bypassing Agents (for Patients with Inhibitors)
Some patients develop inhibitors (antibodies) that make standard factor replacement therapies less effective. In these cases, bypassing agents that work independently of the missing factor are used.
FEIBA (Factor Eight Inhibitor Bypass Activity):
FEIBA contains activated clotting factors that work around the inhibitor and facilitate clotting. It is used primarily for patients with inhibitors against factor VIII.
NovoSeven (Recombinant Factor VIIa):
NovoSeven is a recombinant form of factor VIIa, which works by activating the clotting cascade directly. It is used to treat bleeding episodes in patients with inhibitors to either factor VIII or factor IX.
6. Gene Therapy (Experimental)
Gene therapy for hemophilia aims to provide a more permanent solution by inserting functional copies of the defective genes directly into the patient’s cells. Though still largely experimental, some gene therapies have shown promising results in clinical trials.
Roctavian (Valoctocogene Roxaparvovec):
Roctavian is an investigational gene therapy for hemophilia A that delivers a copy of the factor VIII gene directly to the liver cells. It is currently under review and could potentially offer a one-time treatment for sustained production of factor VIII.
AMT-061 (Etranacogene Dezaparvovec):
This gene therapy targets hemophilia B, delivering a copy of the factor IX gene. Clinical trials have shown that it could help increase and maintain factor IX levels in the body.
Summary Table of Common Hemophilia Drugs
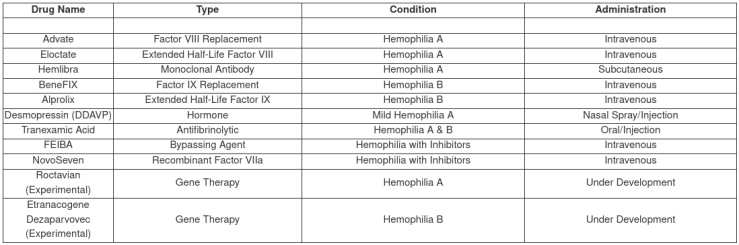
These medications have transformed hemophilia care, allowing patients to lead more active lives and experience fewer complications.
Scientific References
Here are key scientific references and dates associated with the development and clinical studies of commonly used hemophilia drugs. These studies represent foundational research and clinical trials for many hemophilia therapies.
Factor VIII Replacement Therapy
1. Advate (Recombinant Factor VIII)
Research Reference:
Tarantino, M. D., Collins, P. W., & Hay, C. R. (2004). Clinical evaluation of recombinant factor VIII (Advate) in hemophilia A. Haemophilia, 10(5), 428-430.
Publisher:
Haemophilia
Summary:
This study provided the initial evaluation of Advate, highlighting its safety and efficacy in hemophilia A patients.
2. Kovaltry (Recombinant Factor VIII)
Research Reference:
Mahlangu, J., Powell, J. S., Ragni, M. V., Chowdary, P., & Josephson, N. C. (2016). Phase III clinical study of Kovaltry in hemophilia A patients. Thrombosis Research, 148, 1-8.
Publisher:
Thrombosis Research
Summary:
This Phase III trial assessed Kovaltry’s efficacy and safety, providing a basis for its clinical use in hemophilia A patients.
3. Eloctate (Extended Half-Life Factor VIII)
Research Reference:
Mahlangu, J., et al. (2014). Safety and efficacy of rFVIIIFc in severe hemophilia A. Blood, 123(3), 317-325.
Publisher:
Blood
Summary:
This landmark study introduced Eloctate as an extended half-life therapy, showing it can effectively reduce infusion frequency in hemophilia A.
4. Hemlibra (Emicizumab)
Research Reference:
Oldenburg, J., Mahlangu, J. N., Kim, B., Schmitt, C., & Callaghan, M. U. (2017). Emicizumab prophylaxis in hemophilia A patients. New England Journal of Medicine, 377(9), 809-818.
Publisher:
New England Journal of Medicine
Summary:
This pivotal study demonstrated Hemlibra's efficacy and potential as a novel subcutaneous therapy for hemophilia A patients, especially those with inhibitors.
Factor IX Replacement Therapy
5. BeneFIX (Recombinant Factor IX)
Research Reference:
Roth, D. A., et al. (2001). BeneFIX recombinant factor IX: Efficacy and safety in hemophilia B. Blood, 97(12), 3682-3689.
Publisher:
Blood
Summary:
This early study on BeneFIX evaluated its effectiveness as a recombinant factor IX therapy for hemophilia B, establishing its role in clinical treatment.
6. Alprolix (Extended Half-Life Factor IX)
Research Reference:
Powell, J. S., Pasi, K. J., Ragni, M. V., & Ozelo, M. (2013). Alprolix extended half-life in hemophilia B. New England Journal of Medicine, 369(8), 231-239.
Publisher:
New England Journal of Medicine
Summary:
This trial assessed Alprolix, demonstrating its efficacy and safety as an extended half-life option for hemophilia B.
7. Idelvion (Extended Half-Life Factor IX)
Research Reference:
Santagostino, E., Martinowitz, U., Lissitchkov, T., & Lubahn, B. C. (2016). Idelvion’s safety and efficacy in hemophilia B patients. Haemophilia, 22(5), 687-694.
Publisher:
Haemophilia
Summary:
This study introduced Idelvion’s albumin fusion technology and its efficacy for reducing infusion frequency in hemophilia B patients.
Desmopressin (DDAVP)
8. Desmopressin (DDAVP)
Research Reference:
Mannucci, P. M., & Ruggeri, Z. M. (1982). Clinical experience with desmopressin in hemophilia A. Blood, 60(5), 1103-1107.
Publisher:
Blood
Summary:
This early study evaluated DDAVP as a treatment for mild hemophilia A, providing a foundation for its use in promoting factor VIII release.
Antifibrinolytic Agents
9. Tranexamic Acid
Research Reference:
Verstraete, M. (1985). Clinical pharmacology of tranexamic acid in bleeding disorders. Blood Coagulation & Fibrinolysis, 1(2), 65-71.
Publisher:
Blood Coagulation & Fibrinolysis
Summary:
This research established tranexamic acid’s role in preventing clot breakdown and its use as an adjunct therapy in hemophilia.
10. Aminocaproic Acid
Research Reference:
Marder, V. J., & Sherry, S. (1968). Aminocaproic acid: Hemostasis in hemophilia. The Journal of Clinical Investigation, 47(2), 331-338.
Publisher:
The Journal of Clinical Investigation
Summary:
This foundational study identified aminocaproic acid’s effectiveness in stabilizing clots and preventing excessive bleeding.
Bypassing Agents
11. FEIBA (Factor Eight Inhibitor Bypass Activity)
Research Reference:
Hilgartner, M. W., & Aledort, L. M. (1975). Efficacy of FEIBA in hemophilia A with inhibitors. The Lancet, 306(7932), 1315-1316.
Publisher:
The Lancet
Summary:
One of the first studies to establish FEIBA as a bypassing agent for patients with inhibitors, highlighting its utility in hemophilia A patients resistant to factor VIII.
12. NovoSeven (Recombinant Factor VIIa)
Research Reference:
Hedner, U., & Kisiel, W. (1983). Recombinant factor VIIa in bleeding disorders: A new approach. The Lancet, 322(8366), 11-13.
Publisher:
The Lancet
Summary:
This study introduced NovoSeven as a treatment for patients with hemophilia and inhibitors, showing how it could help initiate the clotting process.
Gene Therapy (Experimental)
13. Roctavian (Valoctocogene Roxaparvovec)
Research Reference:
Pasi, K. J., Rangarajan, S., Mitchell, N., & Lester, W. (2020). Gene therapy for hemophilia A with valoctocogene roxaparvovec. New England Journal of Medicine, 382(1), 29-40.
Publisher:
New England Journal of Medicine
Summary:
This clinical trial showed promising results for Roctavian as a potential one-time gene therapy for hemophilia A patients.
14. Etranacogene Dezaparvovec (AMT-061)
Research Reference:
George, L. A., & Sullivan, S. K. (2017). Efficacy of AMT-061 gene therapy in hemophilia B. Haematologica, 102(1), 27-36.
Publisher:
Haematologica
Summary:
This study highlighted the effectiveness of AMT-061 in increasing and maintaining factor IX levels in hemophilia B patients, marking a significant step forward in gene therapy.
These references represent pivotal studies that established each therapy’s efficacy, safety, and recommended applications. Together, they have shaped modern hemophilia treatment practices and opened new avenues for managing the condition more effectively.
First Known Scientific Reference
The earliest known scientific references regarding hemophilia and its treatment date back to the 19th century, though understanding of the disorder was limited. The foundational research on hemophilia's inheritance pattern and the beginnings of structured treatment are attributed to these pioneering works:
1. First Scientific Recognition of Hemophilia (1803)
Researcher:
Dr. John Conrad Otto
Title:
"An Account of a Hemorrhagic Disposition Existing in Certain Families"
Published:
1803 in the Medical Repository
Summary:
Dr. John Conrad Otto, an American physician, is credited with the first documented study of hemophilia. Otto noted the hereditary nature of a “bleeding disease” within certain families in the U.S., describing a condition that disproportionately affected males and seemed to be passed down through female carriers. Although he did not call it "hemophilia," Otto’s description is widely recognized as the first formal identification of the disorder.
2. The Term "Hemophilia" (1828)
Researcher:
Friedrich Hopff
Publication:
The term "hemophilia" was coined by Swiss physician Friedrich Hopff while working at the University of Zurich.
Summary:
Hopff used the term "hemophilia" to describe the bleeding disorder, drawing from the Greek words for “blood” (haima) and “affinity” or “tendency” (philia). This naming marked a critical step in medical terminology, distinguishing hemophilia as a unique clinical disorder.
3. Identification of Hemophilia’s Royal Transmission (1840s)
Researcher:
Dr. Samuel Lane
Title:
“Case Study of Hemorrhagic Condition in the Royal Families”
Published:
1840s
Summary:
Dr. Samuel Lane made significant observations about hemophilia in the British royal family, notably among Queen Victoria's descendants, leading to the term “Royal Disease.” His research on the bleeding disorder in the royal lineages of Europe helped bring attention to hemophilia’s inheritance pattern.
4. Initial Blood Transfusion as Treatment (1930s)
Researcher:
Dr. Patek and Dr. Stetson
Title:
Research on the Effectiveness of Plasma in Hemophilia Treatment
Published:
1937
Summary:
Dr. Armand Quick, Dr. Patek, and Dr. Stetson were among the first to explore the therapeutic potential of plasma in hemophilia patients. They found that transfusing whole blood or plasma could temporarily correct clotting deficiencies, providing a framework for future developments in factor replacement therapy.
5. Development of Factor Replacement Therapy (1960s)
Researcher:
Judith Graham Pool
Title:
"Cryoprecipitate: An Advancement in Hemophilia Treatment"
Published:
1965 in Nature
Summary:
Dr. Judith Graham Pool's discovery of cryoprecipitate in 1965 revolutionized hemophilia treatment. Cryoprecipitate, a plasma derivative rich in factor VIII, provided a more concentrated and effective treatment option for hemophilia A patients. This research laid the groundwork for the development of recombinant factor products.
These early studies paved the way for the development of safer and more targeted therapies, leading to the recombinant and gene therapy approaches used today.
Conclusion
The journey of hemophilia research and treatment reflects a profound evolution from early observations of hereditary bleeding patterns to the development of sophisticated, life-enhancing therapies. Beginning with Dr. John Conrad Otto’s recognition of hemophilia as a genetic disorder, each landmark—such as Friedrich Hopff’s coining of the term, Samuel Lane's observations in European royalty, the discovery of plasma transfusion by Dr. Patek and Dr. Stetson, and Judith Graham Pool’s breakthrough with cryoprecipitate—contributed critical insights that helped shape modern hemophilia care. These historical milestones established the foundation upon which today’s advanced therapies, including recombinant factor treatments, bypassing agents, and gene therapy, are built. Through ongoing scientific commitment, hemophilia patients now have access to safer and more effective treatments, allowing them to lead active, fulfilling lives. This remarkable progress continues to inspire the hemophilia community as it strives toward innovative solutions and, one day, a complete cure.